НАЦИОНАЛЕН СЪВЕТ ПО ЦЕНИ И РЕИМБУРСИРАНЕ НА ЛЕКАРСТВЕНИТЕ ПРОДУКТИ
Наредба за изменение и допълнение на Наредба № 14 от 14 ноември 2019 г. за приемане
на фармако-терапевтично ръководство по ревматология
(обн., ДВ, бр. 94 от 2019 г.; изм. и доп., бр. 38 от 2021 г., бр. 38 и 102 от 2022 г.)
§ 1. В част първа „ВЪЗПАЛИТЕЛНО-СТАВНИ ЗАБОЛЯВАНИЯ“ на приложението към член единствен се правят следните изменения и допълнения:
1.1. В частта „Ревматоиден артрит (по МКБ код М05)“ в т. V „Лекарствена терапия“:
1.1.1. Текстът „Само за tocilizumab и baricitimib има доказателства, че монотерапията е толкова ефективна, колкото комбинация с МТХ.“
се заменя със:
„Само за tocilizumab, baricitimib и upadacitinib има доказателства, че монотерапията е толкова ефективна, колкото комбинация с МТХ.“
1.1.2. В частта „Лекарствени продукти“ т. 3 „Биологични болест-модифициращи антиревматични лекарствени продукти (б-БМАРЛ)“
се изменя така:
„3. Биологични болест-модифициращи антиревматични лекарствени продукти (б-БМАРЛ)
• TNFα инхибитори
– infliximab – химерно човешко-мише моноклонално антитяло от клас IgG1
– etanercept – рецептор за човешки тумор-некротизиращ фактор, p75 Fc фузионен протеин
– adalimumab – рекомбинантно човешко моноклонално антитяло
– certolizumab pegol – пегилиран fab фрагмент от хуманизирано рекомбинантно, моноклонално антитяло, без трансплацентарен трансфер
– golimumab – човешко моноклонално антитяло от клас IgG1κ
• анти-β клетъчна терапия
– rituximab – химерно мише/човешко моноклонално антитяло, представляващо гликозилиран имуноглобулин с човешки IgG1
• инхибитори на ко-стимулацията на Т-клетките
– abatacept – фузионен протеин
• IL-6 рецепторни антагонисти
– tocilizumab – хуманизирано IgG1 моноклонално антитяло срещу човешки интерлевкин-6
• IL-1 рецепторни антагонисти
– anakinra – човешки интерлевкин-1 рецепторен антагонист
Adalimumab е рекомбинантно изцяло човешко IgG1 моноклонално антитяло, насочено срещу TNF-α. Свързва и неутрализира разтворимата и мембранно-свързаната форма на TNF-α. Прилага се подкожно по 40 mg веднъж на 2 седмици. При монотерапия някои пациенти, при които отговорът към терапията с Adalimumab 40 mg през седмица е понижен, могат да имат полза от увеличаване на дозата до 40 mg адалимумаб всяка седмица или 80 mg през седмица. Адалимумаб трябва да се използва по време на бременност само ако е изрично необходимо. Не се очакват ефекти върху кърмените новородени/кърмачета. Следователно Adalimumab може да се използва по време на кърмене.
Adalimumab се прилага за лечение на следните диагнози: М05.0, M05.1, M05.3, M05.8; M08.1, M08.3, M08.4.
Certolizumab pegol е пегилиран fab фрагмент от хуманизирано рекомбинантно, моноклонално антитяло, без трансплацентарен трансфер. Има висок афинитет към човешкия TNF-α както към разтворимата, така и трансмембранната му форма. Не съдържа Fc фрагмент. Препоръчителната начална доза при възрастни пациенти е 400 mg (прилагана като 2 подкожни инжекции по 200 mg дневно) на седмици 0, 2 и 4. След началната доза препоръчителната поддържаща доза е 200 mg на всеки 2 седмици. След като е потвърден клиничен отговор, може да се обмисли алтернативна поддържаща доза от 400 mg на всеки 4 седмици. Понастоящем Certolizumab pegol е първото и единствено одобрено от FDA биологично лекарство за лечение на нерентгенографски axSpA на базата на 52-седмично проучване C-AxSpAnd. (Secukinumab също получи одобрение от FDA за лечение на нерентгенографски axSpA). Перспективно събираните данни от над 500 бременности, с експозиция на Certolizumab pegol, с известен изход на бременността, включващи над 400 бременности, с експозиция през първия триместър, не показват малформативен ефект на Certolizumab pegol. Въпреки това наличният клиничен опит е прекалено ограничен, за да се заключи с разумна сигурност, че няма увеличен риск, свързан с прилагането на Certolizumab pegol по време на бременност. Certolizumab pegol трябва да се използва по време на бременност само при клинична необходимост. CRIB проучване – проспективно, постмаркетингово, фармакокинетично проучване с дизайн за точна оценка на нивото на плацентен трансфер на CZP от майките към децата. CZP е показал липсващ до минимален плацентарен трансфер (<0.1%).
Certolizumab pegol може да се прилага при кърмене, а по време на бременност само при клинична необходимост.
Certolizumab pegol се прилага за лечение на следните диагнози: М05.0, M05.1, M05.3, M05.8.
Etanercept е напълно човешки разтворим TNFα рецептор. Той е димер, състоящ се от извънклетъчната част на два рецептора за TNF, прикрепени към Fc фрагмента на човешкия имуноглобулин G1. Свързва се компетитивно с TNF-α или TNF-α в циркулацията и по този начин блокира тяхното взаимодействие с рецепторите за TNF върху клетъчната повърхност. При възрастни се прилага в доза 25 mg два пъти седмично или 50 mg веднъж седмично подкожно. Etanercept трябва да се използва по време на бременност само при категорична необходимост. Жените с детероден потенциал трябва да обмислят използването на подходяща контрацепция, за да избегнат забременяване по време на терапията с etanercept и в рамките на 3 седмици след прекратяване на терапията.
Etanercept се прилага за лечение на следните диагнози: М05.0, M05.1, M05.3, M05.8; M08.1, M08.3, M08.4.
Golimumаb е рекомбинантно изцяло човешко IgG1моноклонално антитяло, насочено към TNF-α. Моноклоналните антитела свързват в стабилен комплекс както разтворимата, така и трансмембранната форма на TNF-α. Прилага се подкожно по 50 mg веднъж месечно.
При пациенти с телесно тегло над 100 kg, при които след 3 до 4 дози не се постигне задоволителен клиничен отговор, може да се обсъди повишаване на дозата на голимумаб до 100 mg веднъж месечно, като се вземе предвид повишеният риск от определени сериозни нежелани лекарствени реакции при дозата от 100 mg в сравнение с дозата от 50 mg. При пациенти без данни за терапевтичен ефект след 3 до 4 допълнителни дози от 100 mg продължаването на лечението трябва да се преоцени.
Golimumab се прилага за лечение на следните диагнози: М05.0, M05.1, M05.3, M05.8.
Infliximab е химерно моноклонално антитяло, съдържащо човешки и миши компоненти. Свързва се специфично с разтворимата и трансмембранна форма на TNF-α. Препоръчителната доза за лечението на болни с РА е 3 mg/kg, последвана от допълнителни инфузии в доза 3 mg/kg на 2-рата и 6-ата седмица след първата инфузия, след което – на всеки 8 седмици. Infliximab трябва да се прилага едновременно с метотрексат.
Наличните данни показват, че клиничен отговор обикновено се постига до 12 седмици от началото на лечението. При пациенти с недостатъчен отговор или загуба на отговор след края на този период може да се обсъди постепенно повишаване на дозата с приблизително по 1,5 mg/kg до максимална доза 7,5 mg/kg веднъж на всеки 8 седмици. Като алтернатива може да се обсъди и приложение в доза 3 mg/kg на всеки 4 седмици. Ако се постигне добър отговор, лечението на пациента трябва да продължи с избраната доза или при избраните интервали.
Продължаването на лечението трябва да се обмисли внимателно при пациенти, при които няма данни за терапевтично повлияване през първите 12 седмици от лечението или след корекция на дозата.
Infliximab се прилага за лечение на следните диагнози: М05.0, M05.1, M05.3, M05.8.
Tocilizumab е хуманизирано IgG1 моноклонално антитяло срещу човешкия IL-6 рецептор, получено чрез рекомбинантна ДНК технология. RoActemra, в комбинация с метотрексат (MTX), е показан за:
• лечение на тежък, активен и прогресиращ ревматоиден артрит (РА) при възрастни пациенти, които преди това не са лекувани с МТХ;
• лечение на умерен до тежък активен РА при възрастни пациенти, които са се повлияли недостатъчно или са имали непоносимост към предходно лечение с едно или повече болест-модифициращи антиревматични лекарства (БМАРЛ) или към антагонисти на тумор-некротизиращия фактор (ТNF). При тези пациенти Tocilizumab може да се прилага като монотерапия в случай на непоносимост към MTX или когато продължително лечение с MTX не е подходящо.
Доказано е, че Tocilizumab намалява скоростта на прогресия на увреждане на ставите, измерена чрез рентгенография, и подобрява телесната функция, когато се прилага в комбинация с метотрексат. Доказано е, че при непоносимост и когaто продължителното лечение с MTX не е подходящо, tocilizumab е предпочитан биологичен агент за приложение като монотерапия. Препоръчителната доза е 8 mg/kg телесно тегло веднъж на всеки 4 седмици. Прилага се чрез венозна инфузия в продължение на 1 час. Регистрирана е и форма за подкожно приложение – 162 mg веднъж седмично. Tocilizumab се прилага за лечение на следните диагнози: М05.0, M05.1, M05.3, M05.8, M08.2, M08.3, M08.4.
Rituximab е химерно мишо/човешко моноклонално антитяло, насочено срещу CD 20 антигена разположен върху В лимфоцитите. Свързвайки се със CD 20 мембранния протеин rituximab индуцира клетъчна смърт чрез апоптоза. Rituximab в комбинация с метотрексат е показан за лечение на възрастни пациенти с тежък активен ревматоиден артрит, които не се повлияват достатъчно или имат непоносимост към други болест-модифициращи антиревматични лекарства (БМАРЛ), включително един или повече видове терапия с инхибитор на тумор-некротизиращия фактор (TNF).
Доказано е, че Rituximab намалява честотата на прогресия на ставното увреждане, измерено чрез рентгенография, и подобрява физическата функция, когато се прилага в комбинация с метотрексат. Прилага се под формата на две интравенозни инфузии по 1000 mg през 14 дни. След 6 – 12 месеца курсът се повтаря. Преди всяка инфузия се прави премедикация с глюкокортикоид (интравенозно), аналгетик и антихистамин (интрамускулно).
Rituximab се прилага за лечение на следните диагнози: М05.0, M05.1, M05.2, M05.3, M05.8.“
1.1.3. Създава се нова т. 4:
„4. Таргет синтетични БМАРЛ (JAK инхибитори)
tofacitinib – селективен инхибитор на JAK1 и JAK3
baricitinib – селективен инхибитор на JAK1 и JAK2
upadacitinib – селективен и обратим JAK1 инхибитор
filgotinib – селективен и обратим JAK1 инхибитор
Tofacitinib е селективен инхибитор на JAK1 и JAK3, като отслабва сигналите на интерлевкините (IL-2, -4, -6, -7, -9, -15, -21) и интерферони тип I и тип II, което води до модулиране на имунния и възпалителния отговор. Препоръчителната доза при ревматоиден артрит е 5 mg, прилагана два пъти дневно, или 11 mg таблетка с удължено освобождаване, прилагана еднократно дневно.
Пациентите, лекувани с тофацитиниб 5 mg филмирани таблетки два пъти дневно, могат да преминат към тофацитиниб 11 mg таблетки с удължено освобождаване веднъж дневно в деня след последната доза тофацитиниб 5 mg филмирани таблетки.
Пациентите, лекувани с тофацитиниб 11 mg таблетки с удължено освобождаване веднъж дневно, могат да преминат към тофацитиниб 5 mg филмирани таблетки два пъти дневно в деня след последната доза тофацитиниб 11 mg таблетки с удължено освобождаване.
Демонстрирана е фармакокинетична еквивалентност (AUC и Сmax) на тофацитиниб 11 mg таблетки с удължено освобождаване веднъж дневно и тофацитиниб 5 mg филмирани таблетки два пъти дневно.
Tofacitinib в комбинация с метотрексат (MTX) е показан за лечение на умерено тежък до тежък активен РА при възрастни пациенти с недостатъчен отговор или с непоносимост към едно или повече модифициращи болестта антиревматични лекарства. Tofacitinib може да се прилага като монотерапия в случай на непоносимост към MTX или когато лечение с MTX не е подходящо. Препоръчителната доза е 5 mg, прилагана два пъти дневно, или 11 mg таблеткa с удължено освобождаване веднъж дневно.
Монотерапията с tofacitinib е съпоставима с другите терапии по отношение на безопасността и ефeктивността. Комбинираната терапия на тофацитиниб 5 mg филмирани таблетки два пъти дневно и тофацитиниб 11 mg таблетки с удължено освобождаване с к-БМАРЛ, най-вече с methotrexate, показва близки клинични резултати до комбинираните терапии с б-БМАРЛ. Доказателства за персистиране на ефикасността при лечението с тофацитиниб за дългосрочен период до 8 години са предоставени от данни от едно текущо и едно завършено открито, дългосрочно проследяващо проучване. Tofacitinib разполага с данни за безопасност, отчетени от дългосрочно проследяващо клинично проучване с продължителност на наблюдение за период от 9,5 години.
Тофацитиниб е показан за лечение на активен полиартикуларен ювенилен идиопатичен артрит (положителен [RF+] или отрицателен [RF-] за ревматоиден фактор полиартрит и разширен олигоартрит) при пациенти на възраст 2 и повече години с недостатъчен отговор към предходна терапия с DMARD. Тофацитиниб може да се прилага в комбинация с метотрексат (MTX) или като монотерапия в случай на непоносимост към MTX, или когато продължаващото лечение с MTX не е подходящо.
Препоръчителната доза за пациенти с полиартикуларен ювенилен идиопатичен артрит и ювенилен ПсА на възраст две и повече години се базира на следните категории за телесно тегло: 10 < 20 kg прилагат се 3,2 mg (3,2 ml перорален разтвор) два пъти дневно; 20 < 40 kg прилагат се 4 mg (4 ml перорален разтвор) два пъти дневно; ≥ 40 kg прилагат се 5 mg (5 ml перорален разтвор или 5 mg филмирана таблетка) два пъти дневно. Пациенти с тегло < 40 kg, лекувани с тофацитиниб 5 ml перорален разтвор два пъти дневно, може да преминат към тофацитиниб 5 mg филмирани таблетки два пъти дневно. Пациентите с тегло < 40 kg не могат да преминат от тофацитиниб перорален разтвор към тофацитиниб таблетки.
Tofacitinib се прилага за лечение на следните диагнози: М05.0, M05.1, M05.3, M05.8; M08.3, M08.4.
Baricitinib е селективен и обратим инхибитор на Janus киназа (JAK) 1 и JAK2. Показан е за лечение на умерено тежък до тежък активен ревматоиден артрит при възрастни пациенти, които не се повлияват адекватно или имат непоносимост към лечение с едно или повече модифициращи болестта антиревматоидни лекарства. Може да се използва като монотерапия или в комбинация с МТХ. Препоръчителната доза е 4 mg веднъж дневно. Доза от 2 mg веднъж дневно е подходяща за пациенти на възраст ≥ 75 години и може да е подходяща за пациенти с анамнеза за хронични или рецидивиращи инфекции. Доза от 2 mg веднъж дневно може да се има предвид също и за пациенти, постигнали траен контрол на активността на заболяването с 4 mg веднъж дневно и отговарящи на изискванията за постепенно намаляване на дозата. Според EULAR има данни, които показват, че baricitinib може да бъде по-ефективен от TNF-инхибитор. (ref. EULAR 2016).
Във всички проучвания пациентите, лекувани с Baricitinib 4 mg веднъж дневно, имат статистически значимо по-изразен ACR20, ACR50 и ACR70 отговор към 12-ата седмица в сравнение с плацебо, MTX или адалимумаб. Времето до поява на ефект е бързо при различните критерии за резултат, със значимо по-изразен отговор, наблюдаван още на седмица 1. Наблюдавана е продължителна, трайна степен на отговор, с ACR20/50/70 отговори, които се задържат най-малко за 2 години, включително при дългосрочното разширено проучване.
Baricitinib се прилага за лечение на следните диагнози: М05.0, M05.1, M05.3, M05.8.
Upadacitinibе селективен и обратим JAK инхибитор. При изследвания върху човешки клетки упадацитиниб инхибира преференциално сигнализирането чрез JAK1 или JAK1/3 с функционална селективност спрямо цитокиновите рецептори, които сигнализират чрез двойки JAK2.
Upadacitinib е показан за лечение на умерен до тежък активен ревматоиден артрит при възрастни пациенти с недостатъчен отговор или непоносимост към едно или повече модифициращи болестта антиревматични лекарства (DMARD). Upadacitinib може да се използва като монотерапия или в комбинация с метотрексат. Препоръчителната доза упадацитиниб е 15 mg веднъж дневно.
Upadacitinib се прилага за лечение на следните диагнози: М05.0, M05.1, M05.3, M05.8.
Filgotinib е обратим JAK1-селективен инхибитор, който се приема перорално. Filgotinib има добре проучен профил полза-риск, без да засяга JAK2-медиираната хематопоеза или JAK3-медиираната имунна защита.
Оптимизирането на профила на селективността към JAK1 инхибирането позволява използването на дози, които позволяват пикова ефикасност, без да се засягат сигналните пътища на JAK2 и JAK3. Така се избягва ограничаваща дозата токсичност, водеща до анемия или сериозна инфекция.
В биохимични тестове Filgotinib преференциално инхибира активността на JAK1 и показа > 5 пъти по-висока ефикасност на Filgotinib за JAK1 по отношение на JAK2, JAK3 и TYK2.
Filgotinib е показан за лечение на умерен до тежък активен ревматоиден артрит при възрастни пациенти с недостатъчен отговор или непоносимост към едно или повече модифициращи болестта антиревматични лекарства (DMARD). Filgotinib може да се използва като монотерапия или в комбинация с метотрексат.
Filgotinib e показан за перорално приложение. Filgotinib трябва да се приема перорално веднъж дневно със или без храна, като може да се приема по всяко време на деня. Таблетките трябва да се гълтат цели и не трябва да се делят, разтрошават или дъвчат. Препоръчителната доза Filgotinib за възрастни пациенти с ревматоиден артрит е 200 mg веднъж дневно. Препоръчва се начална доза от 100 mg веднъж дневно за пациенти на 75 години и повече, тъй като клиничният опит е ограничен.
Filgotinib се прилага за лечение на следните диагнози: М05.0, M05.1, M05.3, M05.8.“
1.1.4. Досегашните „т. 4 Глюкокортикоиди“ и „т. 5 Имуносупресори“ стават съответно „т. 5 Глюкокортикоиди“ и „т. 6 Имуносупресори“.
1.2. В частта „АНКИЛОЗИРАЩ СПОНДИЛОАРТРИТ (по МКБ код М45)“:
1.2.1. В т. VI „Стратегия на терапията“, в част „Биологични БМАРл“, в т. 6 текстът „Таргет синтетични БМАРЛ – upadacitinib.
Upadacitinib е показан за лечение на активен анкилозиращ спондилит при възрастни пациенти с недостатъчен отговор към конвенционалната терапия.“
се заменя със:
„Таргет синтетични БМАРЛ – upadacitinib и tofacitnib
Upadacitinib и tofacitinib са показани за лечение на активен анкилозиращ спондилит при възрастни пациенти с недостатъчен отговор към конвенционалната терапия.“
1.2.2. В т. VII„Съвременно лечение на АС“:
1.2.2.1. „Фигура 2. „Графично обобщение на препоръките за лечение на АС, базирано на експертното мнение на клиницистите и доказателства от научни изследвания. Болестната прогресия с времето е представена по вертикалата в посока от горе надолу.
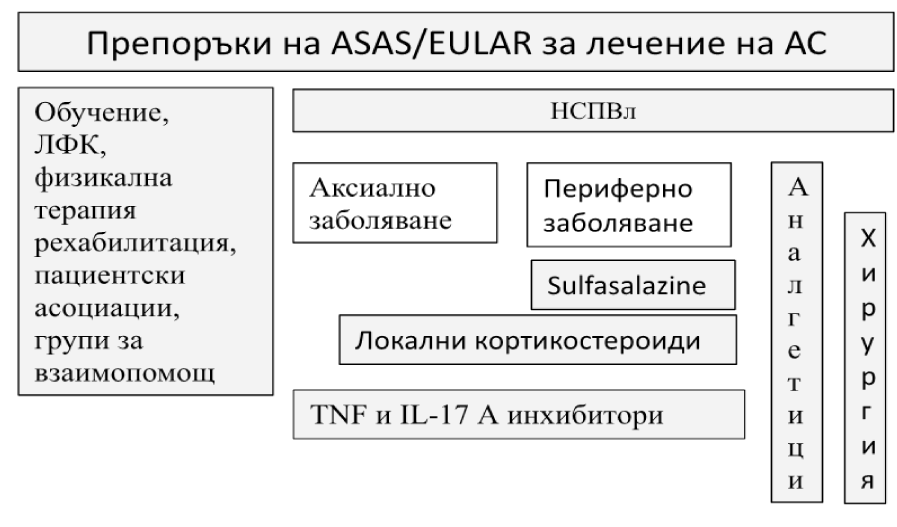
“
се заменя със:
„Фигура 2. „Графично обобщение на препоръките за лечение на АС, базирано на експертното мнение на клиницистите и доказателства от научни изследвания. Болестната прогресия с времето е представена по вертикалата в посока от горе надолу.
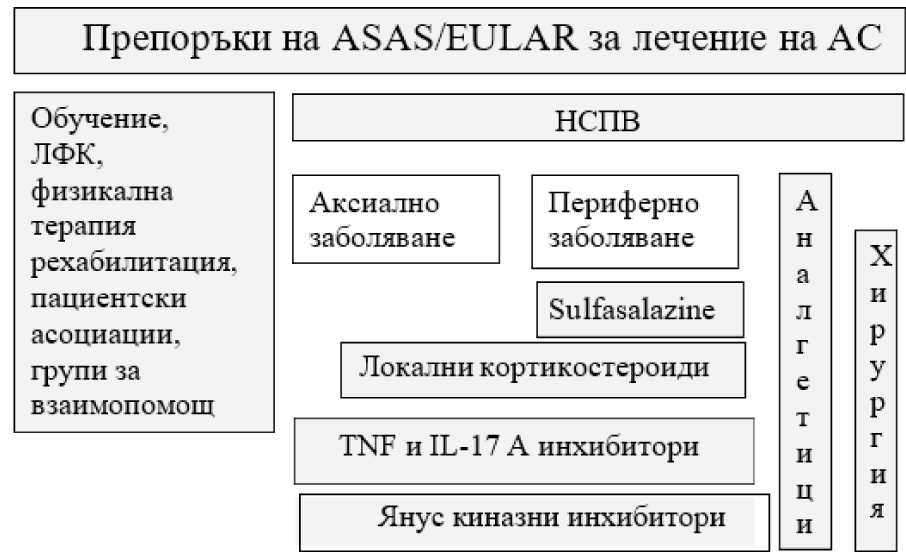
“
1.2.2.2. В „Таблица 4. Преработен вариант на препоръките на ASAS/EULAR за лечение на АС“ се създава нова т. 10:
„10. Таргет синтетични БМАРЛ
- Терапията с таргет синтетични БМАРЛ е с доказана ефективност при активен анкилозиращ спондилит при възрастни пациенти с недостатъчен отговор към конвенционалната терапия.
- Терапията с таргет синтетични БМАРЛ трябва да се има предвид при пациенти с постоянно висока активност на заболяването въпреки конвенционалните лечения.“,
а досегашните т. 10 „Хирургично лечение“ и т. 11 „Промени в хода на болестта“ стават съответно т. 11 „Хирургично лечение“ и т. 12 „Промени в хода на болестта“.
1.2.2.3. В част„Други биологични терапии“,част „IL-17A – блокери“след текста„Upadacitinib се прилага за лечение на следните диагнози: M45.0, M45.1, M45.2, M45.3, M45.4, M45.5, M45.6, M45.7, M45.8.“
на нов ред се добавя:
„Тофацитиниб е показан за лечение на възрастни пациенти с активен анкилозиращ спондилит (АС) с недостатъчен отговор към конвенционалната терапия. Лечението с тофацитиниб 5 mg филмирани таблетки два пъти дневно и тофацитиниб 11 mg таблетка с удължено освобождаване веднъж дневно може да става с преминаване от едната към другата форма в деня след последната доза от съответния вид таблетка. Препоръчителната доза е една таблетка с удължено освобождаване от 11 mg, прилагана веднъж дневно, която не трябва да се превишава. Не се изисква корекция на дозата, когато се използва в комбинация с MTX. Наблюдаваният профил на безопасност при пациентите с активен АС, лекувани с tofacitinib, е сходен с профила на безопасност, наблюдаван при пациентите с РА, лекувани с tofacitinib. Ефективността на tofacitinib при АС е оценена в рандомизирано, двойносляпо, плацебо контролирано клинично проучване, AS-I, с 48-седмично лечение на 269 възрастни пациенти с недостатъчен отговор или непоносимост към най-малко 2 НСПВС. Пациентите, лекувани с тофацитиниб 5 mg два пъти дневно, постигат по-голямо подобрение при ASAS20, и ASAS40 отговорите в сравнение с плацебо на седмица 16. Отговорите се запазват от седмица 16 до седмица 48 при пациентите, получаващи тофацитиниб 5 mg два пъти дневно. Тофацитиниб се прилага за лечение на следните диагнози: M45.0, M45.1, M45.2, M45.3, M45.4, M45.5, M45.6, M45.7, M45.8.“
1.3. В частта„ПСОРИАТИЧЕН АРТРИТ (по МКБ код М07)“:
1.3.1. В т. IV „Стратегия на терапията“:
1.3.1.1. В частта „Биологична БМАРл терапия“:
1.3.1.1.1. точка 6 се изменя така:
„6. Secukinumab самостоятелно или в комбинация с метотрексат (MTX), е показан за лечение на активен псориатичен артрит при възрастни пациенти, при които предшестващата терапия с модифициращи болестта антиревматоидни средства (DMARD) е била недостатъчна.
a. При пациенти с ПсА и съпътстващ умерено тежък до тежък плакатен псориазис или които не са се повлияли достатъчно от проведената анти-TNFα терапия, препоръчителната доза е 300 mg.
b. При TNF – наивни пациенти и пациенти с лек до умерен псориазис препоръчителната доза е 150 mg. Въз основа на клиничния отговор дозата може да се повиши до 300 mg.
c. При недостатъчен клиничен отговор при лечение на псориатичен артрит и тежък псориазис (при пациенти с телесно тегло над 90 кг) се препоръчва приложението на Secukinumab 300 mg през 14 дни. Всяка доза от 300 mg се прилага като една подкожна иннжекция от 300 mg или като две подкожни инжекции от 150 mg.
d. Клиничният отговор обикновено се постига в рамките на 16 седмици лечение.“
1.3.1.1.2. в т. 9 „Guselkumab“, буква b, след текста „Лечението с Guselkumab води до значими подобрения в показателите за болестна активност в сравнение с плацебо на Седмица 24.“
на нов ред се създава нова част „Таргет синтетична БМАРЛ терапия“ със следното съдържание:
„Таргет синтетична БМАРЛ терапия
1. Upadacitinib
a. Упадацитиниб е селективен и обратим инхибитор на Janus киназата (JAK). JAK са вътреклетъчни ензими, които предават сигналите на цитокините или растежните фактори, участващи в широк спектър от клетъчни процеси, включително възпалителни реакции, хемопоеза и имунен отговор. При изследвания върху човешки клетки упадацитиниб инхибира преференциално сигнализирането чрез JAK1 или JAK1/3 с функционална селективност спрямо цитокиновите рецептори, които сигнализират чрез двойки JAK2. Упадацитиниб е показан за лечение на активен псориатичен артрит при възрастни пациенти с недостатъчен отговор или непоносимост към едно или повече модифициращи болестта антиревматични лекарства (DMARD). Упадацитиниб може да се използва като монотерапия или в комбинация с метотрексат.
b. Ефикасността и безопасността на упадацитиниб 15 mg веднъж дневно са оценени в две рандомизирани, двойнослепи, многоцентрови, плацебо контролирани фаза 3 проучвания при пациенти на възраст 18 години или повече с умерен до тежък активен псориатичен артрит. Лечението с 15 mg упадацитиниб води до подобрение при отделните компоненти на ACR, включително броя на чувствителни/болезнени и подути стави, глобални оценки на пациентите и лекарите, HAQ-DI, оценка на болката и hsCRP в сравнение с плацебо. Ефикасността на упадацитиниб 15 mg е доказана независимо от оценените подгрупи, включително изходен BMI, изходен hsCRP и брой на предходните небиологични DMARDs (≤1 или >1).
2. Tofacitinib
a. Tofacitinib в комбинация с MTX е показан за лечение на активен псориатичен артрит (ПсА) при възрастни пациенти с недостатъчен отговор или непоносимост към предходно лечение с модифиращо болестта антиревматично лекарство (DMARD). Препоръчителната доза е 5 mg филмирани таблетки, прилагани два пъти дневно, или една таблетка с удължено освобождаване от 11 mg, прилагана веднъж дневно. Препоръчителната доза не трябва да се превишава. Не се изисква корекция на дозата, когато се използва в комбинация с MTX. Лечението с tofacitinib 5 mg филмирани таблетки два пъти дневно и tofacitinib 11 mg таблетка с удължено освобождаване веднъж дневно или обратно може да става с преминаване от едната към другата форма в деня след последната доза от съответния вид таблетка.
b. Ефикасността и безопасността на tofacitinib филмирани таблетки са оценени в 2 рандомизирани, двойнослепи, плацебо контролирани проучвания фаза 3 при възрастни пациенти с активен ПсА (≥3 подути и ≥3 болезнени стави). В OPAL BROADEN са включени пациенти наивни на биологична терапия, с предходен недостатъчен отговор или непоносимост към csDMARD, а в OPAL BEYOND са включени пациенти, при които е прекратен приемът на инхибитор на TNF поради липса на ефикасност или непоносимост. И в двете проучвания лечението с tofacitinib води до значими подобрения на признаците и симптомите на ПсА, оценено според критериите за ACR20, 50 и 70 отговор. На 3 месец и в двете проучвания се наблюдава значително подобрение на дактилита (LEI) и кожното ангажиране (PASI75). Статистически значимите честоти на ACR20 отговор са наблюдавани при тофацитиниб 5 mg два пъти дневно в двете проучвания още на седмица 2 (първата оценка, извършена след определянето на изходно ниво) в сравнение с плацебо. В проучването OPAL BROADEN, на 12 месец, 96% от пациентите, получаващи tofacitinib 5 mg два пъти дневно, са без рентгенографска прогресия. При пациентите, получаващи tofacitinib 5 mg два пъти дневно и в двете проучвания, се наблюдава подобрение по отношение на артритната болка още на седмица 2 (измереното чрез 0 – 100 визуалната аналогова скала). Наблюдаваният профил на безопасност при пациентите с активен ПсА, лекувани с tofacitinib, е в съответствие с профила на безопасност, наблюдаван при пациентите с РА, лекувани с tofacitinib.“
1.3.2. В т. V „Лечение на ПсА – аксиална ССАП, периферна ССАП, или ПсА. Лечение до постигане на ремисия/ниска болестна активност“:
1.3.2.1. Таблицата след текста „Лечение с лекарствени продукти“се изменя така:
„
|
Лечение
|
Артрит
|
ПсА
|
Спондилит
|
Дактилит
|
Ентезит
|
|
НСПВл
|
X
|
|
X
|
|
|
|
Вътреставно приложение на КС
|
X
|
|
|
|
|
|
Локално лечение (външно)
|
|
X
|
|
|
|
|
Физиотерапия
|
|
|
X
|
|
|
|
Псорален UVA/UVB
|
|
X
|
|
|
|
|
к-БМАРЛ (метoтрексат, циклоспорин A, sulphasalazin, leflunomide)
|
X
|
X
|
|
|
|
|
Билогични БМАРЛ (инхибитори на TNF-α, IL-12/23, IL-17A, IL-23)
|
X
|
X
|
X
|
X
|
X
|
|
Таргет синтетични БМАРЛ (JAK-инхибитори)
|
X
|
X
|
X
|
X
|
X
|
“
1.3.2.2. В частта „Препоръки за лечение на ПсА на БДР от 2019 г., базирани на препоръките на EULAR от 2015 г.“ се правят следните допълнения:
1.3.2.2.1. В т. 5 текстът „Терапията с tofacitinib е показал добра ефективност върху всички прояви на ПсА.“
се заменя с:
„Терапията с tofacitinib или upadacitinib е показалa добра ефективност върху всички прояви на ПсА.“
1.3.2.2.2. В т. 6 след думата „tofacitinib“ се добавят думите„и upadacitinib“.
1.3.2.2.3. В т. 8 след думата „tofacitinib“ се добавят думите„и upadacitinib“.
1.3.2.3. В частта „БМАРЛ (болест-модифициращи антиревматични лекарства)“, точка 2„Таргет специфични болестопроменящи антиревматични лекарствени продукти Инхибитори на JAK“текстът„Лекарствени продукти: Tofacitinib (Xeljanz) – таблетки от 5 мг. Инхибира предимно JAK1 и JAK3. Одобрен е за лечение на ПсА от FDA и EMA в доза 2х5 мг/дн. в комбинация с МТХ или като монотерапия.“ се заличава.
1.3.2.4. В частта „Интерлевкин (IL)-23 инхибитори“:
1.3.2.4.1. След думата „Risankizumab“:
1.3.2.4.1.1. На нов ред се добавя следният текст:
„Ризанкизумаб е хуманизирано имуноглобулин G1 (IgG1) моноклонално антитяло, което селективно се свързва с висок афинитет към p19 субединицата на цитокина човешки интерлевкин 23 (IL-23), без да се свързва с интерлевкин 12 (IL-12), и инхибира взаимодействието му с рецепторния комплекс за IL-23. IL-23 е цитокин, който участва във възпалението и имунните отговори. Чрез блокиране на свързването на IL-23 с неговия рецептор ризанкизумаб инхибира IL-23-зависимата клетъчна сигнализация и освобождаването на проинфламаторни цитокини.
Лекарствена форма: Ризанкизумаб 75 mg инжекционен разтвор в предварително напълнена спринцовка. Разтворът е безцветен до бледожълт и бистър до слабо опалесцентен.“
1.3.2.4.1.2. В частта „Дозировка и начин на приложение:“след текста „Препоръчителната доза е 150 mg, приложени чрез подкожна инжекция на седмица 0, седмица 4 и след това – веднъж на 12 седмици (или като 75 mg предварително напълнена спринцовка – по две инжекции, или като 150 mg предварително напълнена писалка или предварително напълнена спринцовка – по една инжекция).“
се добавя:
„Необходимо е да се обмисли преустановяване на лечението при пациенти, които нямат отговор след лечение в продължение на 16 седмици. Някои пациенти с плакатен псориазис, които първоначално имат частично повлияване, е възможно впоследствие да се подобрят, при продължаване на лечението след 16-ата седмица.“
1.3.2.4.1.3. В частта„Специални популации:“ след текста „Старческа възраст (на възраст 65 или повече години). Не се изисква коригиране на дозата. Има ограничена информация за лица на възраст ≥65 години.“
на нов ред се добавя:
„Педиатрична популация – безопасността и ефикасността на ризанкизумаб при деца и юноши на възраст от 5 до 18 години все още не са установени. Липсват данни.
Няма съответна употреба на ризанкизумаб при деца на възраст под 6 години за показанието умерен до тежък плакатен псориазис или при деца на възраст под 5 години за показанието псориатичен артрит.
Пациенти с наднормено тегло – не се изисква коригиране на дозата.“
1.3.2.4.1.4. След текста „Лечението с Risankizumab води до подобрения по отношение на отделните компоненти на ACR, на Индекса за инвалидност по въпросника за оценка на здравословното състояние (Health Assessment Questionnaire-Disability Index, HAQ-DI), на резултата при оценка на болката и на стойностите на C-реактивния протеин с висока чувствителност (hsCRP) в сравнение с плацебо.“
на нов ред се добавя:
„Клинична ефикасност
Има данни, че при възрастни с активен псориатичен артрит (ПсА), ризанкизумаб подобрява признаците и симптомите, физическата функция, свързаното със здравето качество на живот и процента на участниците без рентгенографска прогресия.
Безопасността и ефикасността на ризанкизумаб са оценени при 1407 участници с активен ПсА в 2 рандомизирани, двойнослепи, плацебо контролирани проучвания (964 в KEEPSAKE1 и 443 в KEEPSAKE2). Участниците в тези проучвания са с диагноза ПсА от най-малко 6 месеца по критериите за класифициране на псориатичен артрит (Classification Criteria for Psoriatic Arthritis, CASPAR), като медианата на продължителността на ПсА е 4,9 години на изходното ниво, имат ≥ 5 болезнени стави, ≥ 5 подути стави и активен плакатен псориазис или псориазис на ноктите на изходно ниво. 55,9 % от участниците имат ≥ 3 % BSA с активен плакатен псориазис. 63,4 % и 27,9 % от участниците имат съответно ентезит и дактилит. В KEEPSAKE1, в което псориазис на ноктите се оценява допълнително, 67,3 % имат псориазис на ноктите. В двете проучвания участниците са рандомизирани да получават ризанкизумаб 150 mg или плацебо на седмици 0, 4 и 16. Като се започне от седмица 28, всички участници получават ризанкизумаб на всеки 12 седмици. В KEEPSAKE1 всички участници са с предходен недостатъчен отговор или с непоносимост към терапия с небиологично DMARD и не са лекувани преди това с биологични средства. В KEEPSAKE2 53,5 % от участниците са с предходен недостатъчен отговор или с непоносимост към терапия с небиологично DMARD и 46,5 % от участниците са с предходен недостатъчен отговор или с непоносимост към биологична терапия. В двете проучвания 59,6 % от участниците са получавали съпътстващо метотрексат (MTX), 11,6 % са получавали съпътстващо небиологични DMARD, различни от MTX, а 28,9 % са получавали ризанкизумаб като монотерапия.“
Заключителна разпоредба
§ 2. Наредбата е приета с Решение на Националния съвет по цени и реимбурсиране на лекарствените продукти, прието съгласно Протокол № 557 от 13.07.2023 г.
Председател: Мила Власковска
5683
